Deciphering Our Genome Using Artificial Intelligence
Every cell in our body contains the same DNA sequence, the same genome. And yet, there is a wide variety of cell types, for example muscle fibers, skin cells, blood cells or even neurons.
In each of these cell types, some genes are expressed, i.e. the corresponding DNA sequence is transformed into RNA and then into proteins, while others are switched off. The instructions that ensure the coordinated expression of these genes during development, and then in the tissue of each organ, are themselves written into the genome.
Only two percent of our DNA sequence codes for proteins. We are currently trying to understand the remaining 98% of the genome, and the coordination of gene expression.
To decipher the rules of this coordination program, we must search in “a book of 3 billion letters”, the equivalent of a novel with a million pages!
This is where artificial intelligence will play an important role, but before we try to understand how, we need to summarize what we know about how this program is implemented.
Locked safes
Initial discoveries showed us that it is the way the DNA molecule folds within the chromosomes that defines the gene regulatory program. A simplified image of this folding would be that of a bank vault.
Each vault contains a gene, and the sequence of that gene is used to make a protein only if that vault is opened. Each safe has a combination lock and only a few combinations are able to open it.
In the below picture, each cell is a replica of the vault, a combination is a set of transcription factors, and for each gene—each vault—there is a unique, cell-specific combination. This combination corresponds to the set of transcription factors present in the cell. These transcription factors activate or inhibit genes by binding to the DNA molecule at specific sites.
Thus, a given combination of transcription factors corresponds to a set of unlocked boxes and a set of expressed genes. In different regions of a developing embryo, these factors may be present or absent, and the response of our genome to their presence or absence enables the appearance of specialized tissues in the desired locations.
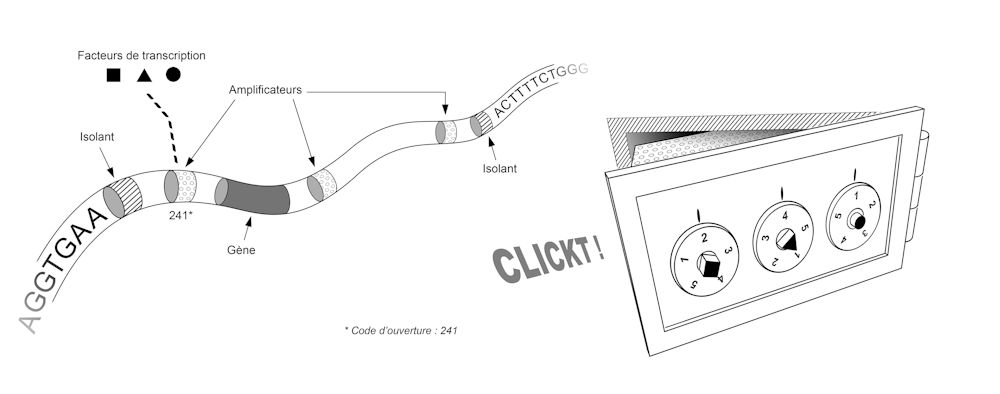
The sequence of our genome thus contains the plan that allows us to build not only the set of vaults, but also the set of locking systems for these vaults. This plan involves two types of elements, each corresponding to small sequences of a few dozen letters (A, C, T or G). First, the "insulators" allow the genome to be partitioned, i.e. to define where each box begins and ends. Then the "amplifiers" are used to make the locking system that validates or invalidates the opening of each box. To get an idea of the size and complexity of this system, imagine that our genome contains about 30,000 genes, a similar number of insulators, and tens or even hundreds of amplifiers for each gene.
Experiments for a better understanding
To improve our understanding of the activity of all these components during development, large-scale experiments are being carried out. These experiments rely on our ability to read, or sequence, the order of letters in the genome.
Sequencing techniques, which were previously only usable on sets of millions of cells, can now be applied to single cells.
These developments make it possible for the first time to simultaneously reveal all regulatory elements (insulators and enhancers) and their activity in different cells over time.
Although research in this field is making great strides thanks to new technologies, one question remains unanswered: how to determine the effect of a genome variation on the gene regulation process?
This question is of crucial importance to understand why certain diseases have a genetic predisposition and thus how to treat an individual better when we know his genetic heritage.
It is commonly observed that certain recurrent variations in the genome can play a role in the appearance or aggravation of diseases. The vast majority of these variations appear in regions of the genome that are not genes, but isolating or amplifying regions.
An algorithm to analyze DNA sequences
To understand the effect of these genome variations, it is now possible to use artificial intelligence. The idea is simple: use all the experimental data obtained so far to train an algorithm to predict the activity of the insulating and amplifying regions according to their genomic sequence.
To do this, we first convert the four letters A, C, T and G into a binary language of 0 and 1. Then we train neural networks similar to those used in image recognition algorithms, used for example to digitize handwritten documents or to analyze images from on-board cameras in autonomous vehicles.
All these applications are based on the same principle: convert a set of numbers, called input, into another set of numbers, called output. This conversion is obtained in several steps. A set of pre-outputs are calculated by multiplying each digit of the input by a coefficient and then by adding the results obtained. This process is repeated by changing the coefficients to generate hundreds or thousands of pre-outputs, which will constitute a layer of the network. The set of pre-outputs of this first layer is used as input to a second layer. Several layers are thus stacked up to the last one, which gives the output of the network. The training process consists in setting the values of the coefficients that make each input correspond to the output. To test these values and optimize them, billions of very simple operations must be performed. This is now possible thanks to the performance of modern graphics cards developed initially for video games.
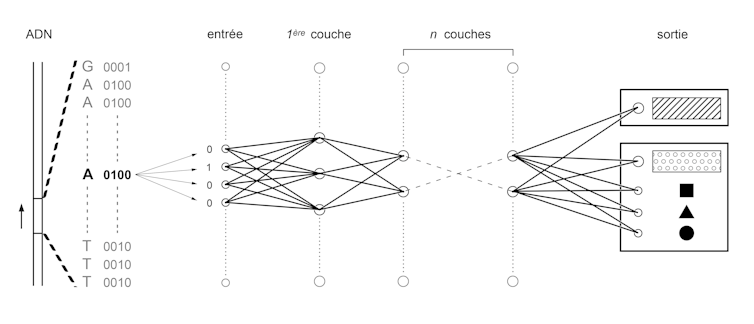
In our case, the input will be a sequence of 0's and 1's corresponding to a DNA sequence in binary form. The output will be another sequence of 0 and 1 that will represent a functional annotation (e.g. 10 or 01 for "amplifier" or "isolator" and 00001000 or 01000000 will correspond to different combinations of transcription factors). Once the algorithm has been trained, it can then be used to predict the functional annotation of a sequence in which one or more letters have been changed: the famous variations. A team of researchers at Princeton University tested genome variations known to be common in people with autism and were able to identify how they modified the combination of genes expressed in brain cells.
The same methodology has been applied to other diseases such as Crohn's disease or chronic hepatitis B infection. In the coming years, personalized medicine should be able to use this methodology to adapt a treatment according to genomic data collected for each individual.
By Julien Mozziconacci, Professor of Computational Biology, Muséum national d'histoire naturelle (MNHN) and Etienne Routhier, PhD student in data science, Sorbonne University
This article is republished from The Conversation under a Creative Commons license (originally in French).